Improving microbiome analysis by deep learning
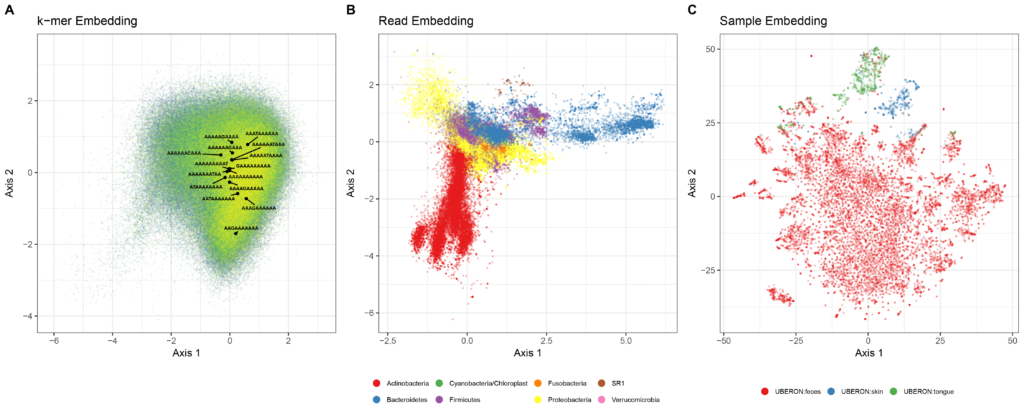
Lower dimensional projections of k-mer, sequence, and sample embeddings. Embedding results were generated using 256 dimensional embeddings of 10-mers that were denoised. A: A 2-dimensional projection via independent component analysis of the 10-mer embedding space from the GreenGenes training sequences. 406,922 unique 10-mers are shown. The position of 10-mers that differ by one nucleotide from AAAAAAAAAA are labeled to demonstrate that it is not simply sequence similarity that is preserved, since these sequences span a wide range in the embedding space. The k-mers were sorted alphabetically and ranked; the alphabetical progression of the indexes are shaded from yellow to green. B: A 2-dimensional projection via independent component analysis. 705,598 total sequences embeddings from 21 randomly chosen American Gut samples (7 from each class) are shown. The position of each sequence (points) is colored based on its phylum designation (only the 7 most abundant phyla are shown). C: 2-dimensional t-SNE projection of the 11,341 American Gut sample embeddings. The position of each sample (points) is colored based on its body site label.
Bacterial traits such as antibiotic resistance (AR)which can have profound impact on their host. These traits are related to genetic variants that can be attributed to larger-scale events (e.g., genetic recombination) or local events (single nucleotide polymorphisms (SNPs)). To understand DNA sequence variation in microbiome samples, local DNA structure should be learned, and to understand how strain variation influences clinically important traits/phenotypes, both local and global (akin to genome-wide association studies (GWAS)). Identifying local genetic variants in bacteria is difficult because bacterial reproduction is clonal and a substantial proportion of the bacterial genome is in strong linkage disequilibrium. Also, it is sometimes difficult to detect large-scale (a.k.a. long-term) events like genetic recombination and horizontal gene transfer (HGT) among lineages. Both local and large-scale features can help resolve genetic variants influencing clinically important phenotype, akin to the genome-wide association studies (GWAS). A comprehensive approach that uses multiple genomic scales is urgently needed to identify meaningful genetic features in the context of phenotype and sample provenance. Doing so will transform our understanding of the role that bacterial genetic variants play in disease.
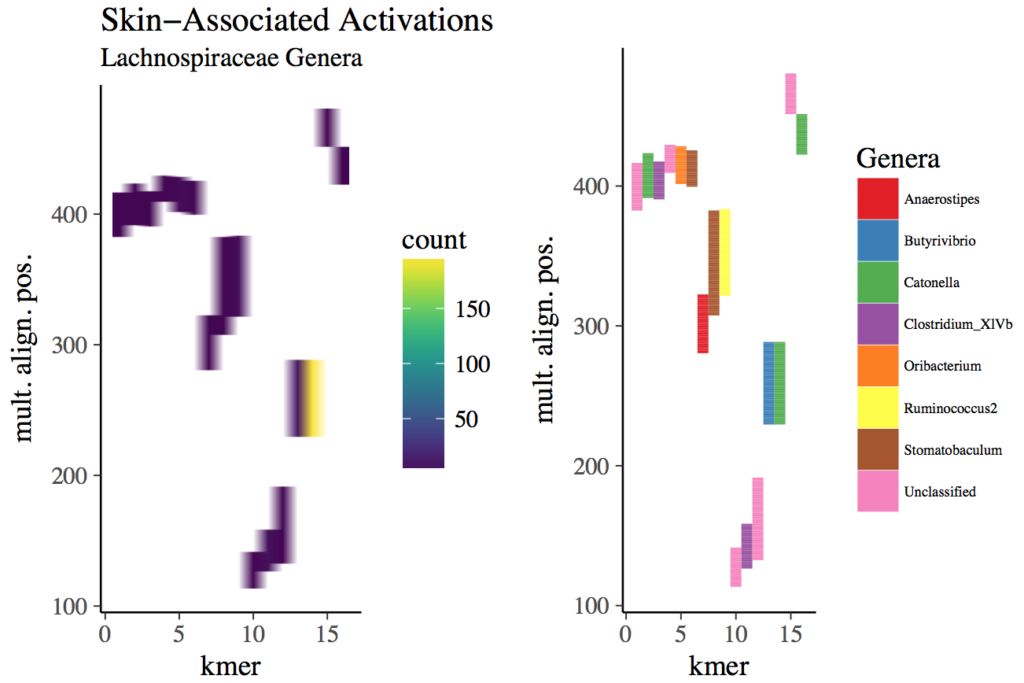
Regions within Lachnospiraceae reads among in which skin-associated k-mers mapped. The top-1000 k-mers with the largest activations (the linear combination of the k-mer embedding and regression coefficients obtained via lasso) for skin were identified. A random sample of 25,000 reads (from skin samples) containing these k-mers underwent multiple alignment. Shown is the relative position of k-mers found only in Lachnospiraceae reads from skin samples. The position of a given k-mer in the alignment spans its entire starting and ending position, including gaps. The frequency in which these k-mers mapped to positions (left column) in the multiple alignment were quantified (y-axis). The order of the k-mers in the heatmap (x-axis) was obtained via hierarchical clustering (Ward’s method) on Bray-Curtis distances. The Lachnospiraceae genus that most frequently occurred at a given alignment position for a given k-mer is colored (right column).
Supervised deep learning can learn the structure of massive data in addition classifying, leading to innovations in many areas. Recent advances in supervised deep learning that leverages a huge volume of data have transferred many research and industrial area to proposing and building novel deep neural networks to learn and infer numerous data they have been collecting so far. Convolutional neural networks (CNNs), Recurrent Neural Networks (RNNs) are very powerful in learning local and global structure of the data. Embedding methods such as word2vec are extremely helpful for deep learning models as they can transform the data into meaningful numerical representations by utilizing large amount of unlabeled data.
(Figures source: Woloszynek, Zhao, Chen, Rosen. PLoS Comp Bio. In Press, 16S rRNA sequence embeddings: Meaningful numeric feature representations of nucleotide sequences that are convenient for downstream analysis. (2019))
Developing an Incremental and Active Learning Framework for Evolving High-Volume Data Streams
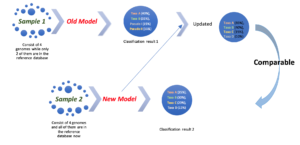
Real-World data is often non-stationary. In addition, as we develop and implement new technology to collect new data, new information can be incompatible with the old datasets. The consequent classification results based on new data are often incomparable with old knowledge due to the change of dimensionality. Hence, we proposed to develop an incremental and active learning framework to address these issues. The proposed framework is able to incrementally update the existing decision boundaries based on new data by a semi-supervised learner. Our framework doesn’t require retraining of the classifier or reprocessing old datasets. To accomplish this goal, we propose to use an incremental semi-supervised learner to update the model and concurrently leverage and update previous unlabeled data. We propose to deliver a software package that can efficiently process a high volume of data that is evolving over time, automatically re-label prior datasets with updated knowledge and leverage large volumes of unlabeled data.
Characterizing the Composition of Metagenomic Samples from Next-Generation Sequencing
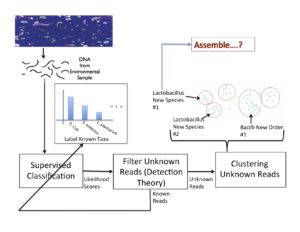
In characterizing communities of organisms from next-generation sequencing, it is important to accurately classify reads to organisms in known databases and then to identify and group novel organisms. Our solution has been to use a supervised classifier in order to maximize and leverage the little information we have in the databases, in order to then predict novel taxa. For the supervised classification problem, we have implemented several methods including support vector machines, cosine similarity and text mining methods, and the Naïve Bayes classifier (NBC) to try to derive an accurate solution. The most fast and accurate solution was the NBC, which we implemented on a website for medical and ecological use: http://nbc.ece.drexel.edu. We also show that detection theory can be used to distinguish between known and novel species and we are investigating ways to now cluster new organisms in an unsupervised manner.
Studying and Comparing the Functional Potential vs. Expression in Biological Communities
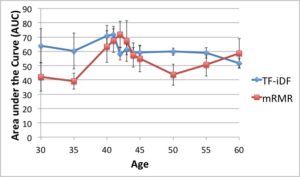
Using the Qin et al., 2010, dataset, we show that age 41/42 gives the best discrimination between two age groups. We believe this signifies a major shift in the microbiome sometime in the early 40’s.
While knowing the taxonomic composition of a sample is important since the community structure may be an indicator of the environment (including disease and other factors), understanding the sample’s function and functional potential is even more useful. Inferring these functions will help us understand these systems and direct design of pharmaceuticals, remediation techniques, etc. to target components in these systems. The EESI Lab is investigating how gene ontology and protein domain relationships can predict environmental conditions. After annotating genes, we classify their function with databases such as Pfam (Protein family database), KEGG (Kyoto Encyclopedia of Genes and Genomes) Pathways database, clusters of orthologous genes (COG) that contain curated families and metabolic pathways of known genes. We are investigating how the occurrence of genes from specific protein families correlate to different human factors such as disease, age, and weight. For example, we have been able to predict if a person had inflammatory bowel disease (IBD) with an accuracy of over 75%. The most significant finding is that the early 40’s yield the best “cut-off” age for predicting whether a person was younger or older than that cut-off measured by the best area-under-the-curve (AUC) of the receiver operating characteristic curve for discriminating the two age groups.
Environmental Community Comparison, Inferring system dynamics, and Modeling Environmental Gradients
Previously, investigators have shown that microbial populations are unique to individuals and that the microbes on a computer keyboard are correlated to the last person to touch it. This is a revolution in forensic analysis — not only can our personal DNA be used in a potential investigation, but the microbes we harbor can reveal our identity. So we ask — can microbes uniquely identify environments? If a certain trace of explosive chemical comes in contact with the soil, how do the microbial populations change with different levels of this chemical? Can we detect the chemical without measuring it directly but through changes in its microbial population? We aim to study microbial populations 16S rRNA gene and whole-genome (and transcriptome) shotgun sequencing to answer these questions.
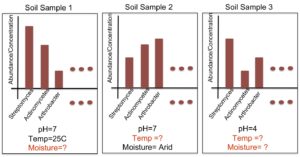
Suppose we have a database of complete taxonomic profiles, each of which has metadata associated it, such as pH, temperature, etc. We then collect and want to analyze more samples to obtain other taxonomic profiles, but for which the metadata might be incomplete (ex: the temperature is missing). This is commonplace in current databases, as very few standards have been implemented about which metadata to collect for which samples; the Genomic Standards Consortium is now leading the way to standardizing such procedures for biologists. So we ask — is there a way to recover the missing parameters from the information available (in order to infer environmental factors from datasets that have been sequenced but ill-labeled)? We aim to answer these questions using function approximation and machine learning prediction. In conjunction with the Dept. of Mathematics and the Biostatistics department, we aim to solve this and other problems such as compressing genomic representation and solving issues with zeros in our data. Out of many problems that the EESI lab works on, this one has most promise to provide ecologists and clinicians with improved data that they may have lost by not recording enough metadata (about environmental conditions).